We collect together here methods to characterize a BigY file. Many are not sure of what they may have downloaded and stored. Made more confusing by the fact that different files were available at different times. Let us start with a rough table and then go from there on how to characterize and recognize what a file is comprised of. There may be manual means of file name patterns mentioned here. But the best, quickest and easiest way is to download and use the tool WGS Extract.
1 Took existing BAMs offline in Fall 2016 while they reprocessed all from HG19 to HG38. No longer made the BigY test or results available, ever again. Only the reprocessed BigY-500 version from the same sequencer run.
2 Coverage as reported by the htslib samtools coverage command; taking into account the >50% 'N' setting in the reference model but not using the ComboBed region to further subdivide (like you would to get WES coverage results from a BAM).
Most other characteristics seem pretty stable otherwise. About 10 million read segments. About 2 gigabases of sequence data. Only mapped reads included. About 70x average read depth estimate. 150 base-pair read segment length. File sizes of anywhere from 700 to 1,400 megabytes. BigY uses the authentic hg19 model including a Yoruba mtDNA model. BigY-500 and onward uses the hg38 model with the rCRS mitochondria reference model. All these characteristics are pretty easily viewed with the WGS Extract Stats button.
The dates are currently best-guess and will be refined over time.
Generating this page has indicated to us that we need a tool, like WES provides for studying the enhanced gene region (Exome), but for the defined ComboBED and similar regions of strongest interest for yDNA. That is clearly what FTDNA enhances for and is not properly characterized in the referenced statistics. So we will work to get the WGS Extract tool enhanced to provide this stat and update the page here once available. Although that may remove the way to distinguish the different files.
Often the original names embedded the aka be shown in the table above.
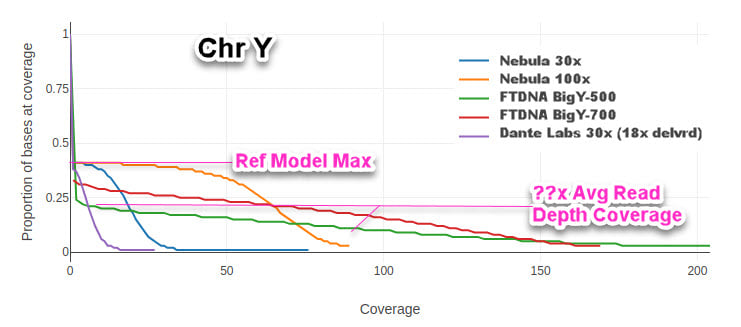
A big distinction is it does not have a sharp rise bell-curve around the average segment length. Instead its slope of the average read depth plotted against the total bases measured is pretty flat horizontally instead of vertical. This can be easily illustrated with a MOSDEPTH plot run on sequencer pair-end segments delivered of various BigY and WGS test results.
| Test (version) | aka | Dates Available | Reference Model | Breadth of Coverage 2 | Notes |
| BigY (v1) | bigy | 2012-2014? | hg19 | 57% +- 5% | Had improperly formatted Qname in every record; included non-Y sequence name entries |
| BigY (v2) | bigy | 2015-2016? | hg19 | 57% +- 5% | Stripped BAM to just Y; removed space in Qname |
| BigY-500 | bigy2 | 2017-2018? | hg38 | 57% +- 5% | Re-processed all existing BigY FASTAs to create Build 38 (not Build 37/19) BAM files identical to new BigY-500 |
| BigY-700 | bigy3 | 2018-present | hg38 | 77% +- 5% | Starting in Nov 2019, all newly purchased tests would no longer provide the BAM file. It has to be separately ordered and paid for. |
2 Coverage as reported by the htslib samtools coverage command; taking into account the >50% 'N' setting in the reference model but not using the ComboBed region to further subdivide (like you would to get WES coverage results from a BAM).
Most other characteristics seem pretty stable otherwise. About 10 million read segments. About 2 gigabases of sequence data. Only mapped reads included. About 70x average read depth estimate. 150 base-pair read segment length. File sizes of anywhere from 700 to 1,400 megabytes. BigY uses the authentic hg19 model including a Yoruba mtDNA model. BigY-500 and onward uses the hg38 model with the rCRS mitochondria reference model. All these characteristics are pretty easily viewed with the WGS Extract Stats button.
The dates are currently best-guess and will be refined over time.
Generating this page has indicated to us that we need a tool, like WES provides for studying the enhanced gene region (Exome), but for the defined ComboBED and similar regions of strongest interest for yDNA. That is clearly what FTDNA enhances for and is not properly characterized in the referenced statistics. So we will work to get the WGS Extract tool enhanced to provide this stat and update the page here once available. Although that may remove the way to distinguish the different files.
File name conventions
First, you have to distinguish between two names. The name of the delivered .zip file. And the name of the BAM and its Index file inside the .zip file.Often the original names embedded the aka be shown in the table above.
Result Characteristics
The BigY test is, similar to WES in that it enriches the sections of the ComboBED region before doing a full sequencing, mapping and variant calling.A big distinction is it does not have a sharp rise bell-curve around the average segment length. Instead its slope of the average read depth plotted against the total bases measured is pretty flat horizontally instead of vertical. This can be easily illustrated with a MOSDEPTH plot run on sequencer pair-end segments delivered of various BigY and WGS test results.
Mosdepth Chart of Y Coverage from same tester
Further Reading
- WGS Extract tool for analyzing BAM files
- Bioinformatics Documents catalogued and written by members of this site
- yDNA Comparison charts (mostly from yFull) of various yDNA analysis (where the MOSDEPTH chart above comes from)